ASD
Atrial Septal Defect (ASD) — A Complete Guide
A clear, thorough guide — from how the heart works to how a hole in the heart is found, treated, and lived with.
ASD at a glance
~1 in 1,500 — live births affected by ASD
2nd — most common congenital heart defect
75% — of ASDs are the secundum type
25% — of adults have a Patent Foramen Ovale (not the same as ASD)
>95% — success rate with device closure
What is an Atrial Septal Defect?
The heart has four chambers — two upper chambers called atria and two lower chambers called ventricles. A wall of muscle called the atrial septum separates the right atrium from the left atrium.
In an Atrial Septal Defect (ASD), there is an abnormal opening (hole) in this wall. Because of this hole, blood that should flow from the left atrium into the left ventricle can “leak” across into the right side of the heart — a situation called a left-to-right shunt. This sends extra blood to the lungs, eventually overloading the right side of the heart.
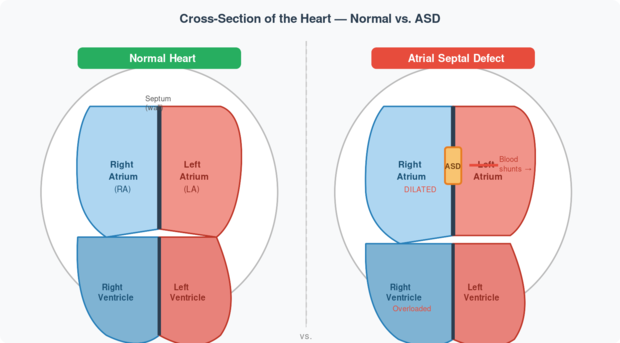
Fig 1. Left: A normal heart with an intact atrial septum. Right: A heart with an ASD — note the hole in the septum (orange), the dilated right atrium, and the left-to-right blood shunt (red arrow). Source: Rudolph’s Pediatric Cardiology, Ch. 8.
💡 Why does blood flow left-to-right? After birth, the left side of the heart has higher pressure than the right (because it pumps blood to the entire body, while the right pumps only to the lungs). This pressure difference pushes blood through the hole from left to right — not the other way around. This is why ASD is called a left-to-right shunt lesion.
Types of Atrial Septal Defect
Fig 2. Classification of atrial-level defects. The secundum ASD accounts for approximately 75% of all cases. Source: Rudolph’s Pediatric Cardiology, Ch. 8; Moss & Adams, Ch. 28.
Why Does ASD Happen?
The atrial septum forms during fetal development around weeks 4–6 of pregnancy. It develops in two overlapping layers — the septum primum and the septum secundum. If these layers don’t form or fuse correctly, a hole remains.
In most cases, no single cause is found. ASD is considered multifactorial — meaning it arises from a mix of genetic susceptibility and environmental factors during pregnancy. Identified associations include:
- Chromosomal syndromes: Down syndrome (Trisomy 21) is strongly associated with primum ASDs and atrioventricular septal defects.
- Single gene mutations: Mutations in the NKX2.5 and GATA4 genes are linked to familial ASD.
- Environmental exposures in pregnancy: Certain medications (e.g., anti-epileptics), alcohol, rubella infection in first trimester.
- Other heart defects: ASD commonly co-occurs with patent ductus arteriosus, ventricular septal defect, or aortic coarctation.
🧠 The Fetal Foramen Ovale — Normal in the Womb. Every baby in the womb has an opening between the atria called the foramen ovale. This is normal and necessary — it allows blood to bypass the non-functioning fetal lungs. After birth, rising left atrial pressure pushes a flap over the opening, sealing it within the first year in ~2/3 of infants. When it doesn’t seal, we call it a Patent Foramen Ovale (PFO) — distinct from an ASD.
Signs & Symptoms
One of the most striking features of ASD is how silent it can be. Unlike ventricular septal defects, even large ASDs rarely cause symptoms in childhood. The right ventricle tolerates the volume overload remarkably well for many years.
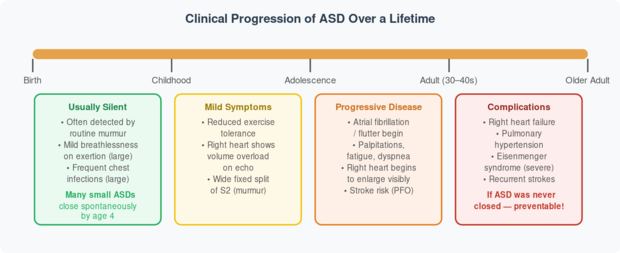
Fig 3. Natural history of an unclosed ASD across a lifespan. Early closure prevents all late complications. Source: Rudolph’s Pediatric Cardiology, Ch. 8.
✅ Classic Doctor’s Finding: The hallmark sign of ASD is a wide, fixed splitting of the second heart sound (S2) that does not change with breathing — unlike in a normal heart. This, combined with a soft systolic ejection murmur at the upper left sternal border, is often how ASD is first suspected during a routine check-up.
How Is ASD Diagnosed?
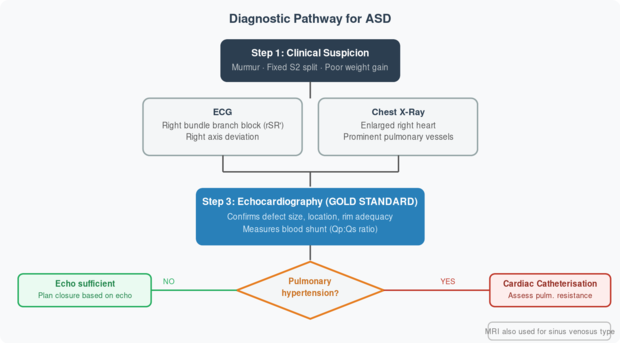
Fig 4. Diagnostic algorithm for ASD. Echocardiography (echo) is the cornerstone. Cardiac catheterisation is reserved for cases with suspected pulmonary hypertension. Source: Moss & Adams, 10th Ed., Ch. 28; Rudolph, Ch. 8.
| Investigation | What It Shows in ASD | Role |
| ECG | Right bundle branch block (rSR’ in V1), right axis deviation, P-wave changes | Screening; suggests right heart overload |
| Chest X-Ray | Enlarged right atrium & ventricle; prominent pulmonary artery; increased lung vascularity | Screening; estimates severity |
| 2D Echocardiogram | Visualises the hole; measures size and rims; shows right heart dilation | Gold standard for diagnosis & planning closure |
| Doppler Echo | Colour flow across the defect; quantifies Qp:Qs (pulmonary:systemic blood flow) | Confirms shunt direction and magnitude |
| TOE (Transoesophageal Echo) | High-resolution images of defect edges (rims) and pulmonary veins | Used before/during device closure |
| Cardiac MRI | Excellent for sinus venosus defects and anomalous pulmonary veins; quantifies RV function | Complex cases; pre-surgical planning |
| Cardiac Catheterisation | Measures pulmonary vascular resistance accurately; oxygen saturations in heart chambers | Reserved for suspected pulmonary hypertension |
Treatment — Closing the Hole
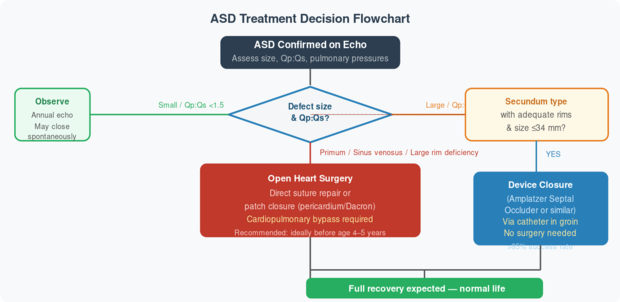
Fig 5. Treatment decision algorithm for ASD. Device closure is preferred when anatomically suitable. Source: Rudolph’s Pediatric Cardiology, Ch. 8; Moss & Adams, Ch. 28.
When should an ASD be closed? The decision to close depends on the size of the shunt (expressed as the ratio of pulmonary-to-systemic blood flow, Qp:Qs). A ratio greater than 2:1 generally warrants closure. Most experts recommend closure before school age (4–5 years) to prevent long-term complications, while the risk of surgery is lowest. Closure after age 40 still reduces arrhythmias and heart failure, though not as dramatically.
🔧 The Amplatzer Septal Occluder is the most widely used device for secundum ASD closure. It is a self-expanding double-disc device made of nitinol wire mesh. Delivered through a thin tube (catheter) inserted in the groin — no chest incision needed. The device straddles the hole on both sides, blocking blood flow, and the body’s own tissue grows over it within a few months. Suitable for defects up to 34–38 mm with adequate surrounding tissue (rims).
Frequently Asked Questions
My child was told they have a “hole in the heart.” Is it serious?
Not always immediately, and often not at all in childhood. Small ASDs may close on their own, and even moderate ones rarely cause symptoms in children. The concern is what happens if the hole remains open into adulthood — then the constant extra blood flow can damage the right heart and lungs. So while it may not feel serious today, it warrants monitoring and, in most cases, closure before it causes problems.
Will an ASD close on its own?
Small secundum ASDs (under 5–6 mm) have a reasonable chance of closing spontaneously, particularly in the first 4 years of life. Larger defects almost never close on their own. Primum, sinus venosus, and coronary sinus defects do not close spontaneously and require intervention. Your cardiologist will advise on how long to observe before acting.
What is the difference between an ASD and a Patent Foramen Ovale (PFO)?
A PFO is a flap-like remnant of the normal fetal circulation that simply didn’t seal after birth — like a door that’s closed but not locked. It exists in about 1 in 4 adults and usually causes no issues. An ASD is a true structural defect — an actual hole where no hole should be. An ASD allows continuous blood flow; a PFO typically only opens under specific pressure conditions (like straining). The main concern with PFO is a slightly elevated risk of cryptogenic stroke from blood clots passing across the flap.
My child’s ASD is being monitored. When will they need treatment?
Treatment is generally recommended if: (1) the defect is causing right heart enlargement or overload on echo; (2) the pulmonary-to-systemic blood flow ratio (Qp:Qs) exceeds 2:1; (3) symptoms develop; or (4) the defect has not closed spontaneously by age 4–5. Most cardiologists recommend closure by school age to allow the right heart to normalise and prevent future arrhythmias.
Is the device (Amplatzer) procedure safe? What are the risks?
Device closure has a success rate exceeding 95% and is now the preferred approach for suitable secundum ASDs. It is a non-surgical procedure — no chest is opened. Risks include: device embolisation (device moving from its position, requiring retrieval), small residual shunts in some patients, and rarely arrhythmias. The Amplatzer device in particular has an excellent long-term safety profile. The patient usually goes home the next day.
Can my child play sports after ASD closure?
Yes, in the vast majority of cases. After successful closure — whether by device or surgery — the right heart normalises over several months. Most children return to full activity, including competitive sports, within 3–6 months. Studies show that exercise capacity returns to normal after closure. Before closure, children with large defects may have mildly reduced exercise tolerance, but most participate in normal activities without restriction.
What happens if ASD is never treated?
If a significant ASD is left unclosed into adulthood, the relentless volume overload of the right heart leads to: atrial fibrillation or flutter (by the 4th–5th decade); right heart failure; pulmonary hypertension (high blood pressure in the lung arteries); and eventually, in severe cases, reversal of the shunt (Eisenmenger syndrome) — a stage where surgery is no longer possible. Early closure prevents all of this completely. This is why doctors recommend closure even in asymptomatic patients.
Will my child need to take medications after closure?
After device closure, antiplatelet medication (usually low-dose aspirin) is given for 6 months while the body’s tissue grows over the device. After surgery, usually no long-term medications are needed. Prophylaxis against infective endocarditis (antibiotics before dental procedures) is no longer routinely recommended for isolated ASDs after the American Heart Association’s 2007 updated guidelines, unless other structural defects co-exist.
Is ASD hereditary? Should siblings or parents be screened?
ASD is not strongly hereditary in most cases — the vast majority occur sporadically. However, familial forms do exist (especially linked to the NKX2.5 gene mutation), and first-degree relatives of a child with ASD have a slightly elevated risk (~3%) compared to the general population. If there is a family history of congenital heart disease, an echocardiogram for siblings is a reasonable precaution but not universally mandated. Genetic counselling may be suggested in familial cases.
Can a woman with ASD have a safe pregnancy?
Most women with a small or repaired ASD tolerate pregnancy well. However, a large unrepaired ASD can worsen during pregnancy because the increased blood volume of pregnancy amplifies the left-to-right shunt, stressing the right heart further. Women with Eisenmenger syndrome face very high maternal and foetal mortality and are advised against pregnancy. Ideally, significant ASDs should be closed before a woman plans to conceive. Women with repaired ASDs generally have normal pregnancies.
🔑 Key Takeaways
ASD is a common, often silent defect. It is the 2nd most common congenital heart defect. Most children have no symptoms — it’s typically found on routine examination via a characteristic heart murmur.
Echocardiography is the diagnostic cornerstone. A 2D Doppler echocardiogram confirms the diagnosis, identifies the type, measures the defect, and guides treatment planning.
Small ASDs may close spontaneously. Defects under 5–6 mm often seal on their own in early childhood. Larger defects require intervention.
Device closure has transformed treatment. The Amplatzer Septal Occluder offers >95% success without open-heart surgery for suitable secundum defects. Primum and sinus venosus types still need surgery.
Early closure prevents all long-term complications. Atrial fibrillation, right heart failure, and pulmonary hypertension are entirely preventable with timely closure — ideally before school age.
Sources
Rudolph AM. Congenital Diseases of the Heart: Clinical-Physiological Considerations, 3rd Ed. Wiley-Blackwell, 2009. Chapter 8: Atrial Septal Defect and Partial Anomalous Drainage of the Pulmonary Veins (pp. 179–202).
Allen HD, Shaddy RE, Penny DJ, Feltes TF, Cetta F (Eds.). Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, 10th Ed. Wolters Kluwer, 2021. Chapters 28, 9, 12, 14.
